
Modeling gas hydrates with the DFTB potential
THEORETICAL CHEMISTRY AND COMPUTATIONAL MODELING

Lab: LCPQ
Duration: NanoX master Internship (8 months part-time in-lab immersion)
5 months full-time internship
6 months full-time internship
Latest starting date: 16/01/2023
Localisation: Laboratoire de Chimie et et Physique Quantiques (LCPQ)
Fédération FeRMI, UMR5626 CNRS & Univ. Toulouse [UT3]
Univ. Paul Sabatier, Bât. 3R4
118 route de Narbonne, Toulouse, France
Supervisors:
Dr Aude Simon aude.simon@irsamc.ups-tlse.fr
Dr Jérôme Cuny jerome.cuny@irsamc.ups-tlse.fr
Work package:
Gas hydrates are a crystalline solid composed of water molecules with cages stabilized by the presence of atoms and molecules. Gas hydrates are present on Earth in the ocean floor and in the permafrost under the form of methane hydrate. They are also suspected to be present on the permafrost of Mars or Titan. This natural occurrence justifies the growing interest for gas hydrates in geoscience and astrophysics. Different forms of hydrates exist depending on the nature of the trapped atoms/molecules that can be rare gas atoms (He, Ne, Ar, Kr) or molecules (H2, N2, O2, CO, CH2, C2H5, C3H8). Understanding the factors that govern the stability of hydrates and their formation is a current significant challenge in chemical physics.
The goal of this internship is, in the framework of a multiscale modeling and in collaboration with experimentalists, to explore the conditions of stability of hydrates of carbon dioxide, cyclopentane, and mix carbon dioxide and cyclopentane, isolated and at the interface with a water surface. The influence of temperature and of the presence of surfactants will be studied. Structural, energetics and dynamic properties such as radial distribution functions will be determined.
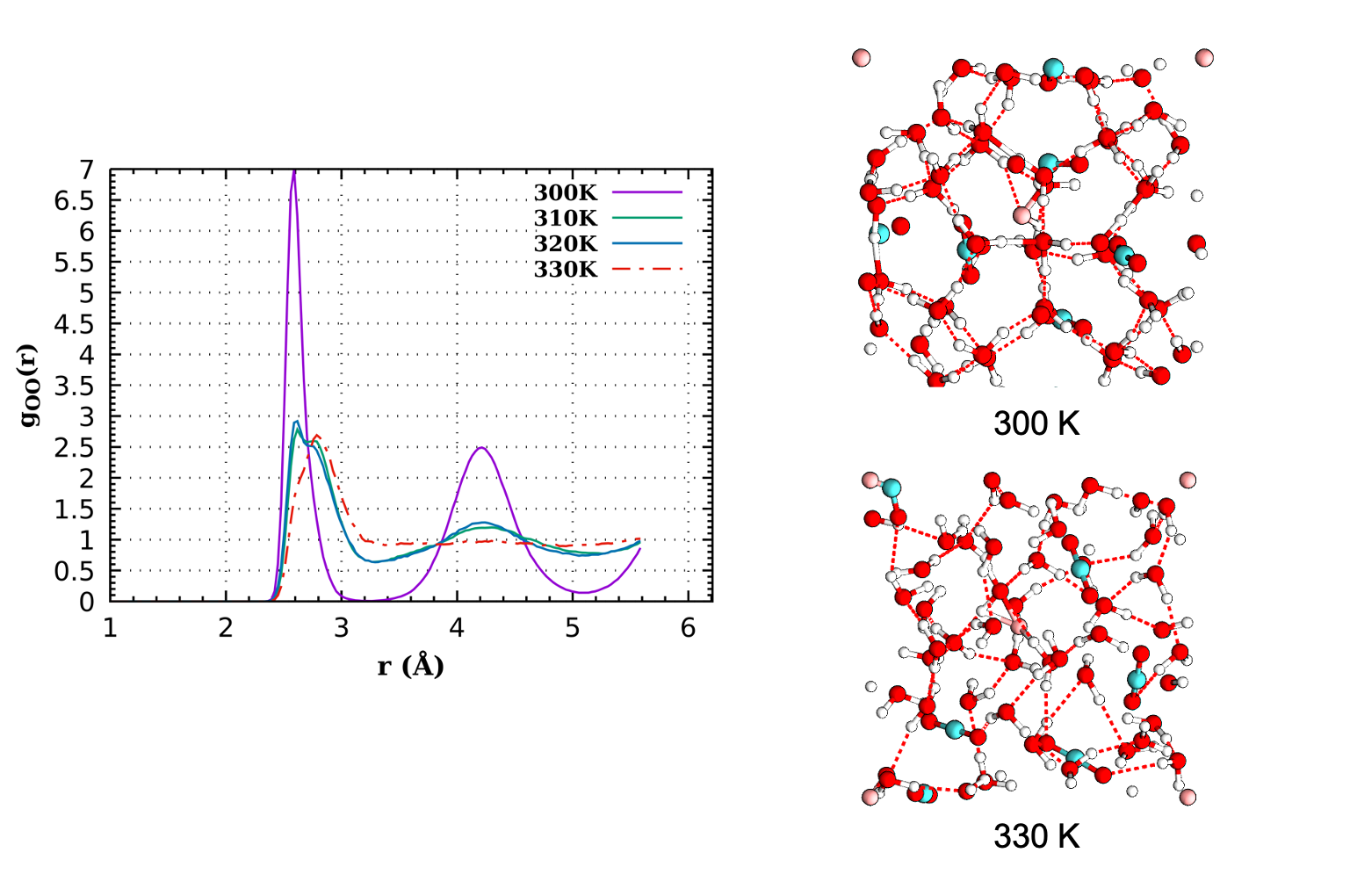
Static calculations and molecular dynamics (MD) simulations will be performed using the density functional based tight binding (DFTB) method with an adjusted potential [1,2]. The DFTB method is an electronic structure method faster than most DFT approaches thus allowing to perform longer simulations. It is slower than force field methods but has the advantage of transferability, which is crucial when chemistry may occur. The internship will follow the PhD by N. Cinq in our group, who worked on the DFTB potential for liquid water [3] and ran the first applications to gas hydrates of CO2 and N2 (see Figure).
References:
[1] F. Spiegelman, N.Tarrat, J. Cuny, L. Dontot, E. Posenitskiy, C. Marti, A. Simon, M. Rapacioli ’Density-functional tight-binding : basic concepts and applications to molecules and clusters’ Adv. Phys. X 2020, 5 :1, 1710252 (10.1080/23746149.2019.1710252, open access)
[2] J. Cuny, J. C. Calatayud, N. Ansari, A. A. Assanali, M. Rapacioli, A. Simon ’Simulation of Liquids with the Tight-Binding Density-Functional Approach and Improved Atomic Charges’, J. Phys. Chem. B 2020, 124, 34, 7421-7432 (10.1021/acs.jpcb.0c04167)
[3] N. Cinq, A. Simon, F. Louisnard, J. Cuny ’An Accurate SCC-DFTB Parametrization of
Liquid Water with Improved Atomic Charges and Iterative Boltzmann Inversion’, J. Phys.
Chem. C 2022, submitted.
Areas of expertise:
quantum chemistry, molecular dynamics simulations
Required skills for the internship:
The trainee must have some general knowledge in quantum chemistry and be motivated to use and adapt computing tools. He will use and elaborate scripts (bash, python, fortran...) to run the quantum chemistry code (the deMonNano suite of programs developped at LCPQ in Fortran90), and to perform post treatment.